Author: Denis Avetisyan
A new method leverages neural networks to dramatically accelerate Markov Chain Monte Carlo simulations, offering a powerful tool for analyzing complex systems and estimating transition probabilities.
This work introduces a neural network-driven importance sampling technique to learn an optimal bias potential, enhancing the efficiency of rare event simulations and improving the accuracy of transition rate estimations.
Simulating the long-term behavior of complex systems is often hampered by the rarity of crucial transition events that dictate material properties. This limitation motivates the development of accelerated simulation techniques, as presented in ‘Accelerated Markov Chain Monte Carlo Simulation via Neural Network-Driven Importance Sampling’, which introduces a novel importance sampling method leveraging neural networks to efficiently explore these rare events. By learning an optimal bias potential, the approach enables accurate estimation of transition rates between metastable states, overcoming the timescale limitations of traditional Markov chain Monte Carlo methods. Could this machine learning-enhanced framework unlock new avenues for simulating complex phenomena across diverse scientific disciplines?
Unveiling Systemic Constraints: The Challenge of Timescales
Atomistic simulations have become indispensable tools for materials science and biophysics, offering insights into material properties by modeling the interactions of individual atoms. However, a fundamental challenge restricts their utility: the timescale problem. Accurately capturing even a fraction of a second of real-world behavior often demands computational times exceeding years, even with the most powerful supercomputers. This limitation stems from the need to solve equations of motion for every atom in the system, requiring a number of computational steps that grows dramatically with both the system size and the desired simulation duration. While algorithms and hardware continue to improve, the inherent complexity of interatomic interactions and the exponential scaling of computational cost with system dimensionality present a persistent bottleneck, forcing researchers to explore approximations and multi-scale modeling techniques to bridge the gap between simulation timescales and relevant physical processes.
Investigating materials systems of even modest size presents a significant hurdle when attempting to model their behavior over timescales relevant to biological processes. The computational demands increase dramatically as researchers strive to simulate dynamic events-like protein folding or cellular signaling-that unfold over milliseconds or even seconds. This intractability doesn’t stem from a lack of algorithmic ingenuity, but rather from the fundamental scaling of computational cost with system complexity. Each additional atom or molecule necessitates a corresponding increase in calculations to determine its interactions and subsequent movements, quickly exhausting available computing resources. Consequently, while simulations excel at revealing atomic-level details, bridging the gap to observe emergent, biologically-relevant phenomena in realistically-sized systems remains a substantial challenge, often necessitating approximations or reduced models that sacrifice some degree of accuracy.
The escalating computational demands of atomistic simulations stem from a fundamental principle: the cost grows exponentially with both the size of the system and the duration of the simulation. This isn’t a linear increase, but rather a multiplicative one; doubling the system’s dimensions, or simulating for twice as long, requires roughly four times the computational power. This phenomenon becomes acutely problematic as researchers move beyond two-dimensional models to investigate the complexities of real-world, three-dimensional materials. The number of interacting atoms, and therefore the calculations needed to predict their behavior over time, increases dramatically, quickly exceeding the capabilities of even the most powerful supercomputers. Consequently, bridging the gap between simulated timescales – often nanoseconds or microseconds – and the biologically relevant timescales of milliseconds, seconds, or even longer, remains a critical challenge in materials science and computational biology.
Accelerating Exploration: Navigating Complex Energy Landscapes
Metadynamics and hyperdynamics are advanced sampling techniques designed to accelerate simulations of infrequent events by actively modifying the potential energy surface. Metadynamics achieves this by iteratively flooding regions of conformational space that have already been visited with Gaussian potentials, discouraging revisits and promoting exploration of new areas. Hyperdynamics, conversely, operates by introducing a history-dependent bias that favors regions of phase space with higher weights, effectively smoothing the energy landscape and increasing the rate of transitions between stable states. Both methods introduce a systematic bias, requiring careful consideration of its impact on the calculation of free energy surfaces and observable properties; appropriate reweighting techniques are often necessary to correct for the introduced bias and obtain accurate results.
Weighted ensemble and forward flux sampling are enhanced sampling techniques designed to improve the efficiency of simulations investigating rare events. Weighted ensemble methods maintain a population of trajectories, collectively exploring the reaction coordinate space, and assign weights to each trajectory based on its contribution to the desired transition. Forward flux sampling, conversely, focuses on identifying and collecting trajectories that successfully cross a defined transition state, iteratively sampling the space until sufficient statistics are gathered. Both methods strategically bias the exploration of reaction pathways, allowing simulations to overcome kinetic bottlenecks and efficiently calculate reaction rates or observe infrequent conformational changes that would be inaccessible via conventional molecular dynamics.
Steered molecular dynamics (SMD) applies external forces to a molecular system to drive it along a predetermined reaction coordinate. This is typically achieved by employing potential energy functions that bias the simulation towards specific configurations, effectively “pulling” or “steering” the system. While SMD accelerates the exploration of relevant conformational space and can provide insights into reaction mechanisms, the applied force inherently introduces bias. The resulting trajectory may not accurately reflect the unbiased dynamics of the system, potentially leading to an overestimation or underestimation of reaction rates and an incomplete representation of the free energy landscape. Careful consideration and validation are therefore necessary when interpreting SMD results.
Molecular dynamics simulations are often limited by the timescale required to observe infrequent, yet critical, events within a system’s potential energy surface. Accelerated sampling methods address this by employing techniques that bias the system’s trajectory, allowing it to overcome energy barriers and explore conformational space more efficiently. These methods do not aim to accurately reproduce equilibrium dynamics, but rather to identify and collect data from relevant configurations – such as transition states or stable minima – that would be rarely visited in standard, unbiased simulations. The collected data can then be used to calculate free energy profiles, reaction rates, or other properties of interest, providing insights into the system’s behavior that would otherwise be inaccessible due to computational constraints.

Refining Statistical Estimates: The Power of Importance Sampling
Importance sampling is a variance reduction technique used in Monte Carlo methods to improve the efficiency of statistical estimations. It achieves this by intentionally oversampling or undersampling regions of the probability space based on their relative importance, assigning weights to each sample to correct for the biased sampling. Specifically, trajectories or configurations with higher contributions to the desired statistical quantity are assigned higher weights, while those with lower contributions receive lower weights. This weighting process ensures that the estimator remains unbiased while reducing its variance, leading to faster convergence and more accurate results with a given number of samples. The efficiency gain is directly proportional to the ability to accurately assess and utilize the relative importance of different regions of the sample space.
Combining importance sampling (IS) with Markov Chain Monte Carlo (MCMC) methods provides a powerful approach for estimating thermodynamic properties by improving sampling efficiency. Standard MCMC simulations can be inefficient when exploring low-probability regions of the phase space; IS addresses this by weighting samples to increase the contribution of these important, yet rare, configurations. This weighting scheme allows for more accurate calculation of ensemble averages, which directly relate to thermodynamic properties such as free energy, pressure, and heat capacity. The combination effectively reduces the variance of the estimator, leading to more robust and precise results, particularly in systems with complex energy landscapes or high dimensionality. The effectiveness of this combined approach depends on the ability to accurately estimate and account for the importance weights during the analysis of the MCMC trajectories.
Effective implementation of importance sampling hinges on precise variance estimation to accurately assess the reliability of weighted samples. Simple variance estimators can be inefficient or biased when dealing with complex probability distributions encountered in molecular simulations or other statistical applications. Consequently, sophisticated statistical estimators, such as those employing jackknife methods, stratified sampling, or control variates, are frequently utilized. These estimators reduce variance and improve the precision of calculated observables, allowing for more efficient convergence and robust statistical inference. Accurate variance estimation is critical for determining appropriate weighting schemes and ensuring the validity of results obtained through importance sampling techniques.
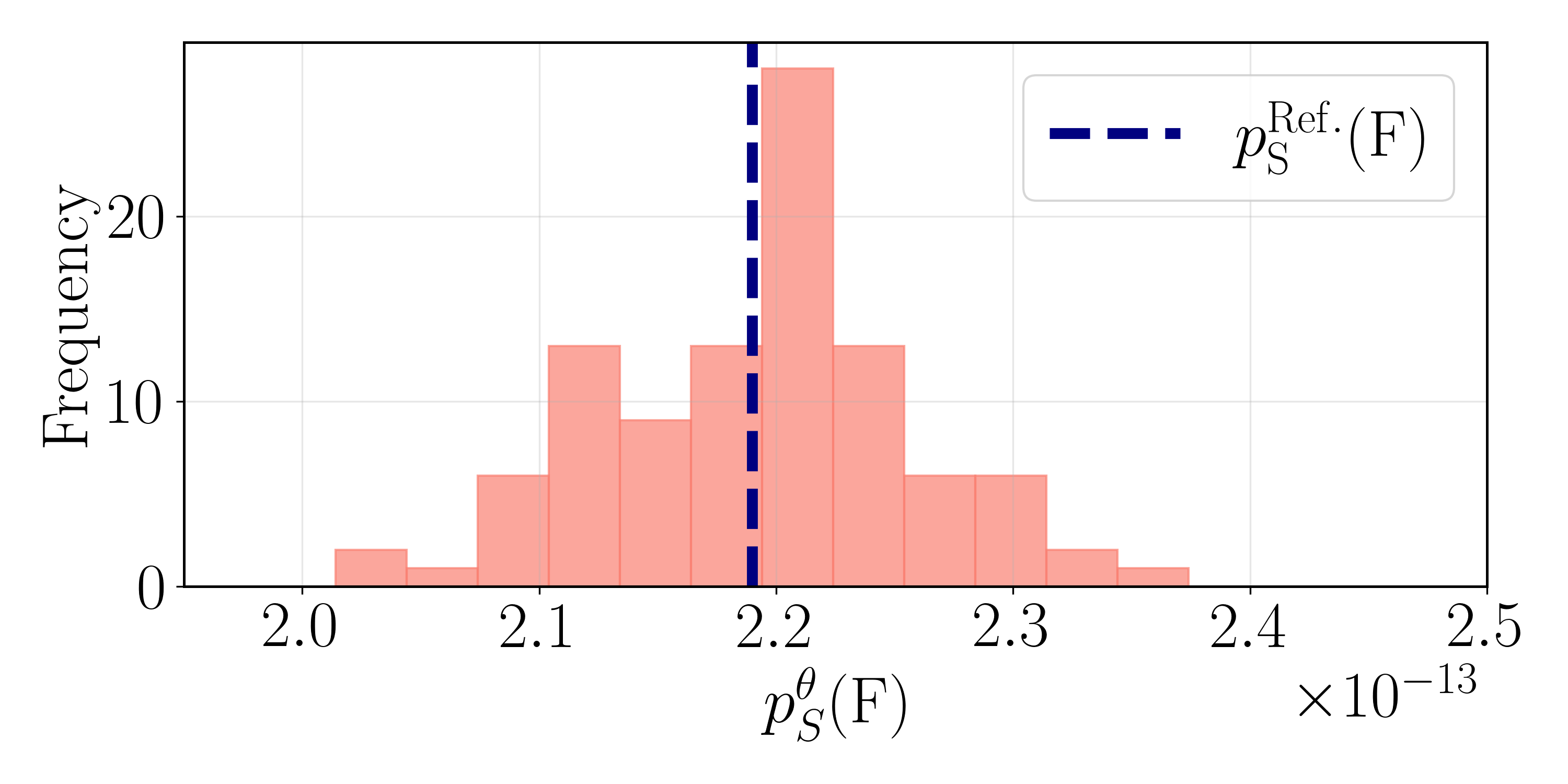
The application of bias potentials in accelerated sampling methods necessitates meticulous attention to maintaining proper normalization to avoid introducing systematic errors. Failure to ensure normalization can lead to inaccurate estimations of free energy landscapes and thermodynamic properties. In a 14-dimensional system examined using these methods, the calculated success probability for a correctly normalized bias potential was determined to be 3.3598 ± 0.0322 x 10-11, indicating the sensitivity of these calculations to even minor deviations from proper normalization procedures and highlighting the need for robust validation techniques.
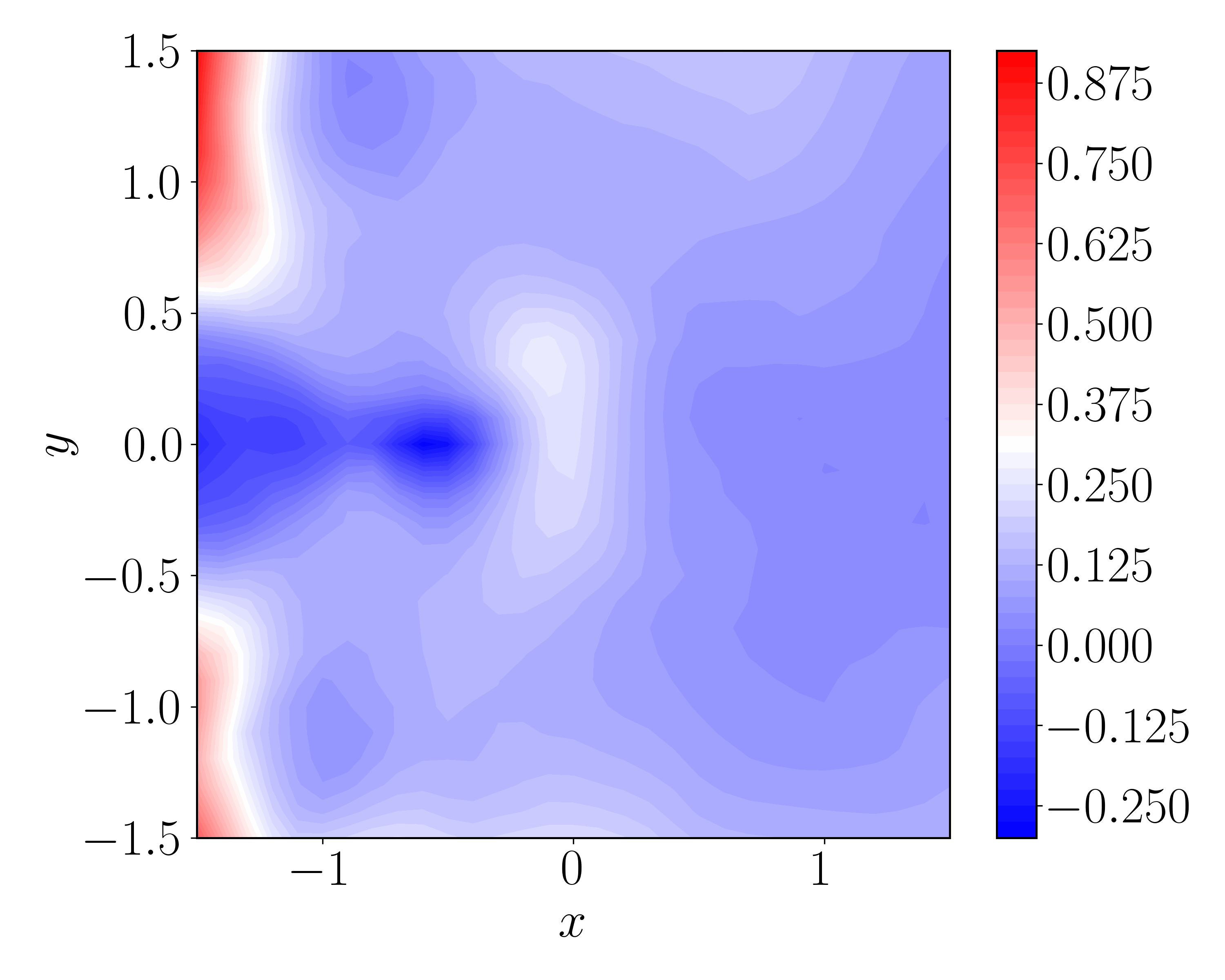
Navigating Computational Artifacts: Ensuring Robustness and Accuracy
Spatially discrete systems, by their nature, rely on approximating continuous phenomena through the use of finite grids. This discretization introduces inherent limitations, as the true, infinitely detailed behavior of a system cannot be perfectly captured by a finite representation. Consequently, any calculated result represents an approximation of the actual physical process; the finer the grid, the more accurate the approximation, but at a significantly increased computational cost. These approximations manifest as errors in quantities like transition rates or probability distributions, demanding careful consideration of grid resolution and the potential impact on overall model fidelity. Researchers must therefore balance computational feasibility with the desire for accuracy, acknowledging that the results obtained from spatially discrete systems are always subject to the limitations imposed by this fundamental approximation.
When simulating systems involving probabilities – particularly those approaching zero – standard computational methods encounter a significant hurdle: numerical underflow. This occurs as computers represent numbers with finite precision, and extremely small values can be rounded down to zero, effectively erasing crucial information about the system’s behavior. To circumvent this, researchers often employ logarithmic space for calculations, transforming probabilities into their logarithmic equivalents. This approach allows for the representation and manipulation of exceedingly small probabilities without succumbing to underflow, as operations performed on logarithms are mathematically equivalent to operations on the original probabilities, but with a dramatically expanded dynamic range. By working with log(P) instead of P, the framework ensures accurate calculations even when dealing with events that have vanishingly small chances of occurring, maintaining the integrity of the simulation and enabling reliable predictions about the system’s state.
The computational demands of simulating complex systems are acutely sensitive to the number of dimensions considered; as dimensionality increases from, for example, a two-dimensional representation to fourteen dimensions, the computational cost escalates dramatically. This is because the state space to be explored grows exponentially with each added dimension, requiring significantly more processing power and memory. However, reducing dimensionality to mitigate these costs introduces approximations that can affect the accuracy of the simulation; a balance must therefore be struck between computational feasibility and representational fidelity. Recent work demonstrates this trade-off by achieving a transition rate of 6.7459 \pm 0.1429 \times 10^{-{12}} in a fourteen-dimensional system, a result that closely aligns with the exact rate of 6.9191 \times 10^{-{12}} obtained from a two-dimensional model, suggesting that, with careful implementation, higher-dimensional simulations can yield remarkably accurate results despite their inherent complexity.
The computational framework demonstrated a remarkable capacity for accuracy even when scaling to high-dimensional systems; specifically, a transition rate of 6.7459 \pm 0.1429 \times 10^{-{12}} was achieved in a fourteen-dimensional system. This result exhibits strong agreement with the precisely calculated rate of 6.9191 \times 10^{-{12}} obtained for the two-dimensional analogue. Furthermore, analysis revealed that 0.2850 of transitions occurred through the S1 state, a value consistent with the theoretical prediction of 0.2938 derived from Kramers’ theory. This level of correspondence validates the framework’s ability to model complex systems and accurately capture key dynamical properties, even as dimensionality increases and computational demands grow.

Towards Predictive Simulations: Shaping the Future of Materials Science
Molecular simulations, crucial for understanding material behavior, often struggle with the ‘time scale bottleneck’ – the inability to observe rare, yet significant, events occurring over extended periods. Researchers are now actively integrating accelerated sampling methods, which intelligently navigate complex energy landscapes to prioritize observation of these events, with robust statistical estimators designed to minimize bias and error. However, simply speeding up the simulation isn’t enough; careful attention to numerical limitations – such as those imposed by computer precision and the discretization of physical space – is paramount. By meticulously addressing these limitations alongside advancements in sampling and statistical analysis, scientists are striving to create simulations capable of accurately predicting long-term material properties and unlocking insights previously inaccessible due to computational constraints. This combined approach promises to dramatically extend the reach of molecular simulations, enabling the study of processes ranging from protein folding to the nucleation of new materials.
Predicting the speed of chemical reactions within intricate systems is often hampered by the energy barriers separating reactants and products; however, Kramers’ rate theory provides a powerful framework for overcoming this challenge. This theory doesn’t require detailed knowledge of the transition pathway, instead focusing on the height and width of the energy barrier, alongside the system’s frictional forces. By characterizing these features, researchers can accurately estimate reaction rates – even in scenarios with multiple competing pathways or complex environmental effects. This approach moves beyond simplistic models, enabling the prediction of reaction dynamics in crowded molecular environments, biological systems, and materials science applications where traditional methods fall short. Consequently, Kramers’ rate theory serves as a cornerstone for understanding and ultimately controlling chemical processes at the molecular level, offering insights crucial for designing new catalysts and materials.
The pursuit of increasingly accurate and efficient molecular simulations hinges on continuous innovation in adaptive sampling strategies and algorithmic design. Current research focuses on methods that dynamically adjust the simulation’s focus, concentrating computational effort on regions of phase space that contribute most significantly to desired properties or events – effectively ‘learning’ where to sample most effectively. This contrasts with traditional methods that explore the entire space uniformly, often wasting resources on irrelevant configurations. Advanced algorithms, including those employing machine learning techniques, are being developed to accelerate these sampling processes and refine statistical estimators, minimizing computational cost while maintaining or improving the precision of results. Such advancements promise to unlock simulations of larger, more complex systems, and over longer timescales, ultimately providing deeper insights into material behavior and accelerating the design of novel materials.
The convergence of increasingly sophisticated simulation techniques promises a revolution in materials science and engineering. By accurately modeling the behavior of matter at the atomic and molecular level, researchers can now virtually design materials with pre-defined characteristics – from enhanced strength and conductivity to tailored optical and catalytic properties. This capability bypasses the traditionally slow and expensive trial-and-error approach to materials discovery, enabling the rapid identification of promising candidates for applications ranging from energy storage and conversion to biomedical implants and advanced electronics. The ability to predict material performance before synthesis not only accelerates innovation but also minimizes resource waste and allows for the creation of sustainable, high-performance materials optimized for specific challenges.
The pursuit of efficient simulation, as detailed in this work, mirrors the core of scientific inquiry – discerning signal from noise. It’s akin to refining a microscope’s lens to bring rare phenomena into clearer view. Igor Tamm observed, “The universe is not governed by blind chance, but by a complex interplay of laws and probabilities.” This sentiment perfectly encapsulates the paper’s approach. By leveraging neural networks to learn an optimal bias potential for importance sampling, the researchers effectively ‘tune’ the simulation, amplifying the probability of observing rare events – much like enhancing a weak signal. The ability to accurately estimate transition rates, a key outcome of this method, allows for a deeper understanding of the underlying dynamics governing complex systems, revealing patterns previously obscured by computational limitations.
Future Directions
The pursuit of efficient rare event simulation inevitably reveals the limitations of any single methodological approach. This work, while demonstrating the power of neural network-driven importance sampling, merely shifts the challenge-from exploring the state space to accurately representing the bias potential with a limited training dataset. The inherent tension between exploration and exploitation remains, and future efforts will likely focus on adaptive learning strategies-methods that dynamically adjust the bias potential during the simulation itself. One can anticipate a branching of approaches: those seeking increasingly sophisticated neural network architectures, and those revisiting the fundamental principles of importance sampling to reduce reliance on learned parameters.
A curious direction lies in extending this framework beyond static bias potentials. Complex systems rarely remain static; their transition rates and underlying dynamics evolve. Incorporating temporal dependencies into the neural network-treating the bias potential as a function of time, or even a stochastic process itself-could unlock simulations of non-equilibrium phenomena currently beyond reach. This raises intriguing questions about the very definition of ‘importance’ in a dynamic context.
Ultimately, the success of such methods will be judged not by computational speed alone, but by the ability to reveal previously hidden patterns in complex systems. The pursuit of efficiency is, after all, merely a means to an end: a clearer understanding of the underlying rules governing the world-a world which, one suspects, will continue to resist complete comprehension.
Original article: https://arxiv.org/pdf/2602.12294.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- United Airlines can now kick passengers off flights and ban them for not using headphones
- Katanire’s Yae Miko Cosplay: Genshin Impact Masterpiece
- All Itzaland Animal Locations in Infinity Nikki
- How to Complete Bloom of Tranquility Challenge in Infinity Nikki
- How to Get to the Undercoast in Esoteric Ebb
- Gold Rate Forecast
- A Dark Scream Theory Rewrites the Only Movie to Break the 2-Killer Rule
- All Golden Ball Locations in Yakuza Kiwami 3 & Dark Ties
- BloxStrike codes (March 2026)
- Fire Force Season 3 Part 2 Episode 24 Release Date, Time, Where to Watch
2026-02-17 03:42