Author: Denis Avetisyan
Researchers have developed a framework that moves beyond analyzing genes in isolation to predict how cells will react to genetic changes by modeling coordinated gene programs.
scBIG leverages module-inductive bias and flow matching to generate more accurate predictions of transcriptional responses to single-cell perturbations.
Predicting cellular responses to genetic perturbations remains a significant challenge, often hindered by methods that treat genes as independent entities despite evidence of coordinated transcriptional programs. To address this, we present scBIG, a framework detailed in ‘Beyond Independent Genes: Learning Module-Inductive Representations for Gene Perturbation Prediction’, which explicitly models these interconnected gene programs through a module-inductive bias and conditional flow matching. This approach enables more accurate and generalizable predictions of perturbation responses, consistently outperforming existing methods-with an average improvement of 6.7%-particularly in challenging combinatorial settings. Can leveraging structured biological priors unlock even more robust and interpretable models for understanding gene regulatory networks?
Decoding Cellular Response: The System’s Hidden Language
The ability to accurately predict cellular responses to genetic alterations – known as gene perturbation – represents a cornerstone of modern biomedical research. This understanding isn’t merely academic; it’s fundamentally linked to advancements in both disease modeling and the rational design of therapeutic interventions. By systematically altering gene expression, researchers can mimic the molecular hallmarks of disease, creating in vitro and in vivo models to investigate disease mechanisms and test potential drug candidates. Moreover, a comprehensive grasp of how cells respond to these perturbations allows for the identification of novel drug targets and the prediction of drug efficacy, ultimately accelerating the development of personalized medicine approaches tailored to an individual’s unique genetic profile. The precision with which scientists can anticipate these responses directly impacts the success rate of drug discovery and the effectiveness of future therapies.
Predicting how a cell’s gene expression shifts following a genetic alteration – a process known as transcriptional response – proves remarkably difficult for conventional analytical approaches. These methods often focus on individual genes, failing to capture the intricate, coordinated changes occurring across entire functional groups of genes. This simplification overlooks the fact that genes rarely act in isolation; instead, they participate in complex regulatory networks where the effect of a perturbation on one gene can ripple through numerous others. Consequently, traditional techniques frequently underestimate the full scope of the cellular response and generate inaccurate predictions, hindering progress in both disease modeling and the development of targeted therapies. Capturing the dynamic interplay between genes, rather than treating them as independent units, is therefore paramount for a more complete and accurate understanding of cellular behavior.
Current approaches to understanding cellular responses often dissect gene perturbation by examining individual gene expression changes, overlooking the intricate, coordinated activity of functional gene programs. This isolated view presents a significant limitation, as genes rarely act in isolation; instead, they operate within networks where the expression of one gene profoundly influences others. Researchers are discovering that a more holistic approach – one that maps how perturbations ripple through these interconnected programs – is crucial for accurate predictions. Capturing these coordinated changes requires advanced computational models and experimental techniques capable of resolving not just which genes change, but how their expression patterns shift in relation to one another, ultimately revealing a more complete picture of cellular behavior and opening new avenues for targeted therapies.
SCBIG: Reverse-Engineering Cellular Programs
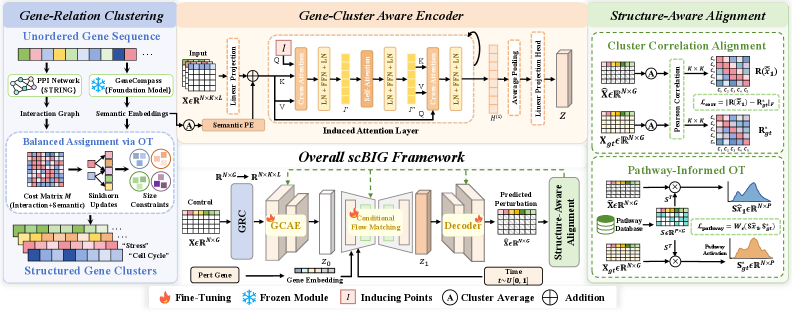
SCBIG functions as a generative framework for predicting transcriptional responses by directly modeling the coordinated activity of gene programs. Unlike methods focused on individual gene predictions, SCBIG aims to capture the relationships between genes operating as functional modules. This is achieved through a process of explicitly representing these coordinated programs, allowing the model to forecast how entire groups of genes will respond to cellular stimuli. The generative nature of the framework implies it doesn’t just predict expression levels, but rather simulates the underlying biological processes driving transcriptional change, offering a more holistic and mechanistically informed prediction of cellular behavior.
The SCBIG framework employs a gene-cluster-aware encoder to represent cellular states and model relationships between gene programs. This encoder functions by initially grouping genes into clusters based on shared functional characteristics, thereby establishing a higher-level representation of the genome. Induced attention mechanisms are then integrated, allowing the model to dynamically weigh the importance of different gene clusters when representing a given cellular state. This process captures inter-module dependencies by enabling information flow between these pre-defined gene clusters, moving beyond individual gene-level analysis and focusing on coordinated transcriptional responses. The resulting encoded representation effectively captures the complex interplay between gene programs, providing a more nuanced understanding of cellular behavior.
SCBIG incorporates a module-level inductive bias through architectural constraints and training objectives designed to prioritize the learning of coordinated gene program responses. This is achieved by explicitly modeling relationships between groups of genes – termed modules – rather than treating each gene independently. The model is encouraged to predict changes in module activity based on input perturbations, effectively learning how entire programs of genes respond in a coordinated fashion. This contrasts with traditional approaches that focus on individual gene predictions, and facilitates generalization to unseen conditions by leveraging the inherent structure of gene regulation.
Traditional gene expression prediction models often operate on a gene-by-gene basis, establishing direct relationships between individual genes and external stimuli. SCBIG diverges from this approach by explicitly modeling coordinated gene programs, recognizing that cellular responses are rarely driven by isolated gene activations. Instead of predicting the expression of each gene independently, SCBIG focuses on predicting changes at the level of functional modules – groups of genes that work together to execute specific biological processes. This module-centric approach allows SCBIG to capture complex interdependencies and generalize more effectively to unseen conditions, as it learns to predict program-level responses rather than relying on individual gene-to-gene mappings. Consequently, SCBIG is capable of identifying and predicting coordinated changes in gene expression that would be missed by models focusing solely on individual genes.
Ensuring Biological Consistency: Constraining the Simulation
SCBIG utilizes Pathway-informed Optimal Transport as a biological consistency objective to improve the accuracy of its predictions by ensuring alignment with established biological pathways. This method leverages known pathway structures to constrain the model’s generative process, effectively reducing the likelihood of biologically implausible predictions. Specifically, Optimal Transport is employed to minimize the distance between predicted gene expression changes and those observed in known pathway responses to perturbations. This constraint is implemented by defining a cost function that penalizes deviations from expected pathway-driven expression patterns, thereby promoting outputs consistent with current biological understanding and reducing false positive predictions.
Cluster Correlation Alignment (CCA) is a regularization technique employed within SCBIG to maintain the expected relationships between gene expression modules during both training and inference. This is achieved by explicitly minimizing the divergence between the correlation structure of gene expression in the predicted perturbed state and that of the control state. CCA leverages the principle that functionally related genes tend to exhibit coordinated expression patterns; therefore, preserving these module co-expression patterns – representing inherent functional gene programs – improves the biological plausibility of SCBIG’s predictions and enhances its ability to accurately model cellular responses to perturbation. The method operates on pre-defined gene clusters representing these modules, enforcing consistency in their relative expression levels rather than individual gene predictions.
SCBIG utilizes a conditional flow matching backbone to model the transitions observed between control and perturbed cellular states. This approach frames the generative modeling process as learning a continuous flow that maps noise to data, conditioned on the specific perturbation applied. Unlike diffusion models which require numerous iterative denoising steps, flow matching directly learns the velocity field guiding the data distribution, resulting in faster sampling and improved robustness. The conditional aspect enables the model to generate realistic responses to various perturbations by learning distinct flow trajectories for each condition, effectively capturing the underlying biological mechanisms driving the observed changes in gene expression.
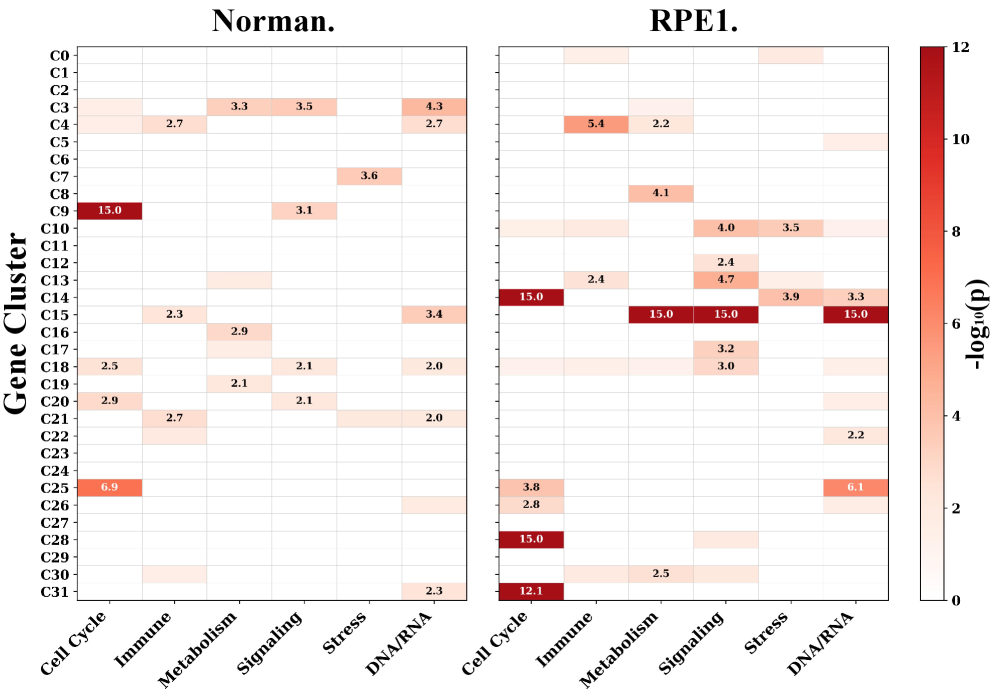
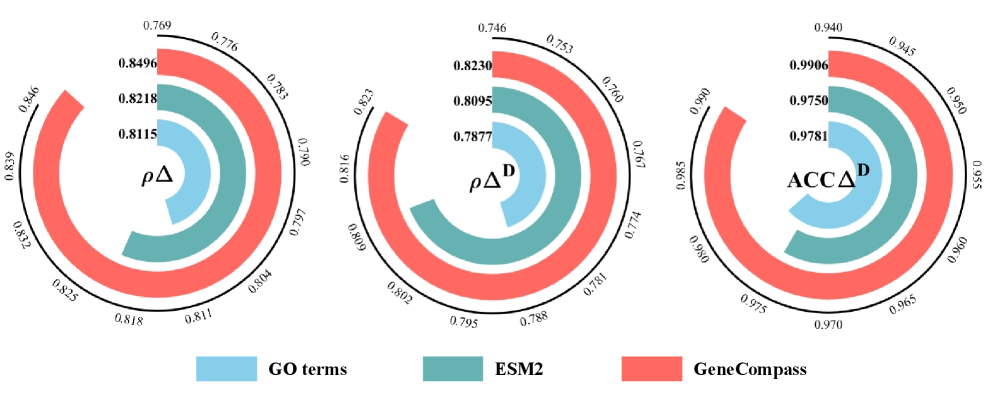
SCBIG utilizes gene-relation clustering to define functional modules, a process driven by both foundation models and established protein-protein interaction (PPI) networks. This clustering approach integrates information from pre-trained models – capturing complex gene relationships learned from large-scale datasets – with the empirically-validated physical and functional associations detailed in PPI networks. The resulting modules represent groups of genes that exhibit coordinated behavior, effectively capturing the inherent organization of cellular processes and providing a foundational framework for subsequent predictive modeling within SCBIG. These modules are not predefined but are dynamically identified based on the input data and the integrated knowledge from foundation models and PPI data.
Benchmarking and Future Horizons: Beyond Prediction
Rigorous validation of SCBIG’s predictive capabilities was achieved through comprehensive testing on established benchmark datasets, notably the Norman Dataset and the RPE1 Dataset. These datasets, widely used within the single-cell RNA sequencing field, provided a standardized platform for comparing SCBIG’s performance against existing methodologies. Utilizing these benchmarks ensured that observed improvements weren’t simply the result of dataset-specific characteristics, but rather indicative of a genuinely enhanced ability to model cellular responses.
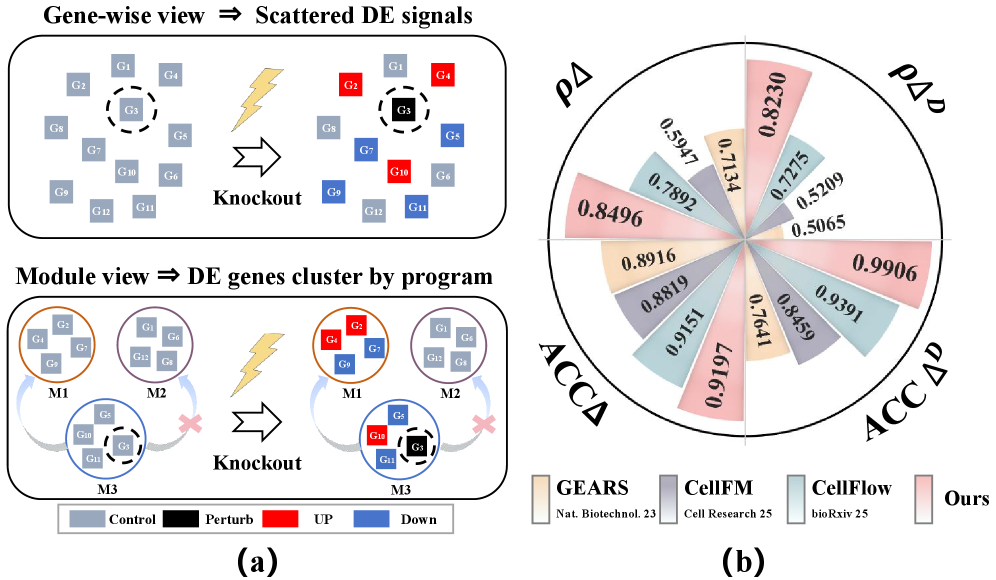
SCBIG establishes a notable advancement in predicting how cells respond to stimuli by demonstrably improving the accuracy of transcriptional response predictions. Rigorous testing reveals an average performance gain of 6.7% when contrasted with existing methodologies, suggesting a heightened capacity to model complex cellular behaviors. This enhancement isn’t merely incremental; the framework’s ability to accurately forecast transcriptional changes promises to refine biological models and, crucially, offers a more reliable foundation for downstream analyses in areas like drug response prediction and the dissection of disease mechanisms. The consistent gains observed across multiple metrics highlight SCBIG’s robustness and potential to become a standard tool for researchers investigating gene expression dynamics.
Rigorous testing reveals that SCBIG demonstrates substantial improvements in predictive accuracy when contrasted with the established CellFlow methodology. On the Norman dataset, SCBIG achieved a 7.7% increase in Pearson Correlation ρΔ, indicating a stronger relationship between predicted and observed transcriptional responses. Even more pronounced gains were observed using the RPE1 dataset, where SCBIG boosted Accuracy Delta ACCΔ by an impressive 34.9% compared to CellFlow. These results collectively suggest that SCBIG offers a significantly enhanced capability for modeling cellular behavior and discerning the effects of perturbations, potentially accelerating advancements in areas like drug response prediction and personalized therapeutic strategies.
Leveraging the power of single-cell RNA sequencing, this technology offers a compelling pathway toward more efficient drug discovery and tailored medical interventions. By precisely charting cellular responses to various stimuli, researchers can identify promising drug candidates with greater accuracy and speed, potentially reducing the lengthy and costly trial-and-error processes traditionally associated with pharmaceutical development. Furthermore, the granular detail afforded by single-cell analysis facilitates a deeper understanding of individual patient variability, opening doors to personalized treatment strategies where therapies are customized based on a person’s unique molecular profile. This capability extends beyond reactive treatment, offering the potential for proactive, preventative medicine designed to address health risks before they manifest, ultimately reshaping the landscape of healthcare through precision and individualization.
Analysis reveals that SCBIG not only predicts transcriptional responses with heightened accuracy, but also excels at discerning the effects of cellular perturbations. Specifically, the framework achieved a superior Perturbation Discrimination Score (PDS) on the RPE1 dataset, indicating a refined ability to differentiate between cells undergoing varying treatments or stimuli. This improvement extends to broader statistical significance, as demonstrated by substantially enhanced Spearman Significance (DES) values observed across both the Norman and RPE1 datasets; these findings suggest that SCBIG’s predictions are more robust and less likely attributable to random chance, thereby strengthening its potential as a reliable tool for understanding complex biological systems and informing targeted interventions.
The pursuit of predictive modeling in biological systems necessitates a dismantling of simplistic assumptions. This work, introducing scBIG, exemplifies that principle; it doesn’t merely accept genes as independent units, but actively seeks the underlying, coordinated programs governing cellular response. As Hannah Arendt observed, “The banality of evil lies in the acceptance of clichés and the unwillingness to think critically.” Similarly, accepting genes as isolated entities is a form of intellectual complacency. scBIG, by incorporating a module-inductive bias and leveraging generative modeling, challenges this ‘banality’ – it reverse-engineers the complexity of gene regulatory networks, seeking not just correlation but causal structure, and thus offers a more robust prediction of transcriptional response to perturbation.
Decoding the Blueprint
The scBIG framework represents a step, not a destination. It acknowledges a fundamental truth: genes don’t act in isolation. They operate as distributed processing units within larger programs-modules-and predicting cellular behavior requires understanding those programs, not merely cataloging individual components. However, this approach implicitly assumes a degree of modularity that may not universally hold. The elegance of the ‘module-inductive bias’ rests on the premise that these modules are, in fact, meaningfully separable-a claim that demands rigorous testing beyond predictive accuracy. Future work must explore the limitations of this assumption and investigate the degree to which gene networks represent genuinely discrete functional units, or instead, a more fluid, interconnected web.
Currently, the method leans heavily on pre-existing biological knowledge to define these modules. This is pragmatic, but ultimately limiting. The real challenge lies in discovering these modules de novo, inferring structure directly from data, and building models that can adapt as new information emerges. It’s a shift from reverse-engineering a known circuit to prospecting for undiscovered functionality. The underlying reality is open source-the code exists-but pinpointing it requires more than just improved algorithms; it demands a willingness to challenge existing interpretations and embrace unexpected connections.
Ultimately, the predictive power of any such framework is bounded by the quality and completeness of the data. Single-cell perturbation studies are inherently complex and expensive, creating an inevitable trade-off between resolution and scale. Future investigations should focus on developing methods that can effectively leverage limited data, perhaps through transfer learning or by integrating information from multiple modalities. The goal isn’t just to predict what a cell will do, but to understand why-to map the logic of life at a resolution sufficient to truly manipulate its code.
Original article: https://arxiv.org/pdf/2602.04901.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- Netflix’s Little House On The Prairie Reboot: Release Date, Cast & Everything We Know
- Off Campus Season 1 Soundtrack Guide
- Silver Rate Forecast
- Prime Video’s New R-Rated Spy Thriller Is Officially No.1 On Streaming Despite Poor Reviews
- Gold Rate Forecast
- Brent Oil Forecast
- Infinity Nikki Candlelight Reverie Challenge and Rewards Guide
- EUR ZAR PREDICTION
- Bulgakov’s Take: Koreans Bet the Farm on Chips, Crypto, and Chaos
- Inkford Hermitage Chest Locations In HSR (Honkai: Star Rail)
2026-02-08 20:59